Thrombozytopenische Purpura bei Kindern ICD-Code 10. Immunthrombozytopenie bei Kindern
Angeborene Thrombozytopenien sind meist Teil erblicher Syndrome wie dem Wiskot-Aldrich-Syndrom, der Fanconi-Anämie, dem Bernard-Soulier-Syndrom, der May-Hegglin-Anomalie usw. Da bei erblichen Thrombozytopenien in der Regel auch qualitative Veränderungen der Blutplättchen beobachtet werden, sind dies der Fall werden üblicherweise als Thrombozytopathien klassifiziert.
Die Ursachen einer erworbenen Thrombozytopenie sind äußerst vielfältig. So kann der Ersatz des Blutverlustes durch Infusionsmedien, Plasma und rote Blutkörperchen zu einer Abnahme der Thrombozytenkonzentration um 20–25 % und dem Auftreten einer sogenannten Verdünnungsthrombozytopenie führen. Die Verteilungsthrombozytopenie beruht auf der Sequestrierung von Blutplättchen in der Milz oder in Gefäßtumoren – Hämangiomen – mit dem Ausschluss einer erheblichen Menge an Blutplättchenmasse aus dem allgemeinen Blutkreislauf. Eine Verteilungsthrombozytopenie kann sich bei Krankheiten entwickeln, die mit einer massiven Splenomegalie einhergehen: Lymphome, Sarkoidose, portale Hypertonie, Milztuberkulose, Alkoholismus, Morbus Gaucher, Felty-Syndrom usw.
Die zahlreichste Gruppe besteht aus Thrombozytopenien, die durch eine erhöhte Zerstörung von Blutplättchen verursacht werden. Sie können sich sowohl im Zusammenhang mit der mechanischen Zerstörung von Blutplättchen (z. B. bei Herzklappenprothesen, künstlichem Kreislauf, paroxysmaler nächtlicher Hämoglobinurie) als auch bei Vorhandensein einer Immunkomponente entwickeln.
Eine alloimmune Thrombozytopenie kann durch die Transfusion von Fremdblut verursacht werden; Transimmun – Eindringen mütterlicher Antikörper gegen Blutplättchen durch die Plazenta zum Fötus. Autoimmune Thrombozytopenie ist mit der Produktion von Antikörpern gegen die eigenen unveränderten Blutplättchenantigene verbunden, die bei idiopathischer thrombozytopenischer Purpura, systemischem Lupus erythematodes, Autoimmunthyreoiditis, multiplem Myelom, chronischer Hepatitis, HIV-Infektion usw. auftreten.
Heteroimmune Thrombozytopenie wird durch die Bildung von Antikörpern gegen fremde Antigene verursacht, die auf der Oberfläche von Blutplättchen fixiert sind (Medikamente, Viren usw.). Eine medikamenteninduzierte Pathologie tritt bei der Einnahme von Beruhigungsmitteln, Antibiotika, Sulfonamid-Medikamenten, Alkaloiden, Goldverbindungen, Wismut, Heparin-Injektionen usw. auf. Nach Virusinfektionen (Adenovirus-Infektion, Influenza, Windpocken, Röteln, Masern, infektiöse Mononukleose), Impfungen.
Eine Thrombozytopenie, die durch eine unzureichende Bildung von Blutplättchen (produktiv) verursacht wird, entwickelt sich mit einem Mangel an hämatopoetischen Stammzellen. Dieser Zustand ist typisch für aplastische Anämie, akute Leukämie, Myelofibrose und Myelosklerose, Tumormetastasen im Knochenmark, Mangel an Eisen, Folsäure und Vitamin B12, die Auswirkungen von Strahlentherapie und zytostatischer Chemotherapie.
Schließlich entsteht eine Verbrauchsthrombozytopenie aufgrund eines erhöhten Bedarfs an Blutplättchen zur Gewährleistung der Blutgerinnung, beispielsweise beim DIC-Syndrom und bei Thrombosen.
RCHR (Republikanisches Zentrum für Gesundheitsentwicklung des Gesundheitsministeriums der Republik Kasachstan)
Version: Klinische Protokolle des Gesundheitsministeriums der Republik Kasachstan – 2016
Idiopathische thrombozytopenische Purpura (D69.3)
Kinderonkologie, Pädiatrie
allgemeine Informationen
Kurzbeschreibung
Genehmigt
Gemeinsame Kommission für Gesundheitsqualität
Ministerium für Gesundheit und soziale Entwicklung der Republik Kasachstan
vom 29. November 2016
Protokoll Nr. 16
Immunthrombozytopenie- eine Autoimmunerkrankung, die durch eine isolierte Thrombozytopenie (weniger als 100.000/μl) mit einer konstanten/erhöhten Anzahl von Megakaryozyten im Knochenmark und dem Vorhandensein von Anti-Thrombozyten-Antikörpern auf der Oberfläche der Blutplättchen und im Plasma der Patienten gekennzeichnet ist, die normalerweise auf die Membran wirken Glykoproteinkomplexe IIb/IIIa und/oder GPIb/IX, was zur Zerstörung von Blutplättchen durch Zellen des phagozytischen mononukleären Zellsystems führt, was sich in einem hämorrhagischen Syndrom manifestiert.
Korrelation von ICD-10- und ICD-9-Codes
| ICD-10 | ICD-9 | ||
| Code | Name | Code | Name |
| D69.3 | Immunthrombozytopenie | - | - |
Datum der Entwicklung des Protokolls: 2016
Protokollbenutzer: Allgemeinmediziner, Therapeuten, Kardiologen, Hämatologen, Kinderärzte, Onkologen.
Evidenzgradskala
| A | Eine hochwertige Metaanalyse, eine systematische Überprüfung von RCTs oder großen RCTs mit einer sehr geringen Wahrscheinlichkeit (++) einer Verzerrung, deren Ergebnisse auf eine geeignete Population verallgemeinert werden können. |
| IN | Hochwertige (++) systematische Überprüfung von Kohorten- oder Fallkontrollstudien oder hochwertige (++) Kohorten- oder Fallkontrollstudien mit sehr geringem Verzerrungsrisiko oder RCTs mit niedrigem (+) Verzerrungsrisiko Die Ergebnisse können auf eine geeignete Population übertragen werden. |
| MIT |
Kohorten- oder Fallkontrollstudie oder kontrollierte Studie ohne Randomisierung mit geringem Verzerrungsrisiko (+). Deren Ergebnisse können auf die relevante Population oder RCTs mit sehr geringem oder niedrigem Risiko einer Verzerrung (++ oder +) verallgemeinert werden, deren Ergebnisse nicht direkt auf die relevante Population verallgemeinert werden können. |
| D | Fallserie oder unkontrollierte Studie oder Expertenmeinung. |
Einstufung
EinstufungAmerikanische Gesellschaft für Hämatologie, 2013:
Mit der Strömung:
· neu identifiziert – Dauer bis zu 3 Monate;
· anhaltende (langwierige) ITP – Dauer 3–12 Monate;
Chronische ITP – Dauer mehr als 12 Monate.
Je nach Schweregrad des hämorrhagischen Syndroms:
· schwer – Patienten mit klinisch signifikanten Blutungen, unabhängig von der Thrombozytenzahl. Fälle, die zu Beginn der Krankheit mit Blutungssymptomen einhergehen und den Beginn einer Therapie erforderlich machen, oder Fälle, in denen die Blutung erneut auftritt und zusätzliche therapeutische Wirkungen mit verschiedenen Arzneimitteln, die die Anzahl der Blutplättchen erhöhen, oder eine Erhöhung der Dosierung des Blutplättchens erforderlich ist Drogen verwendet.
· refraktär – keine oder keine vollständige Reaktion (Blutplättchen unter 30 x 109/l) auf die Therapie nach Splenektomie; Reaktionsverlust nach Splenektomie und die Notwendigkeit einer medizinischen Behandlung zur Minimierung klinisch signifikanter Blutungen. In diesem Fall ist eine erneute Untersuchung erforderlich, um andere Ursachen einer Thrombozytopenie auszuschließen und die Diagnose einer ITP zu bestätigen. Wird hauptsächlich bei Erwachsenen gefunden.
Von Stufen; Standardisierung von ITP, September 2006 IMBACH]:

Diagnostik (Ambulanz)
Ambulante Diagnostik
Diagnosekriterien: ACHTUNG! Eine primäre Immunthrombozytopenie wird diagnostiziert, wenn die Thrombozytenzahl auf weniger als 100 x 109/l absinkt, wobei andere Ursachen der Thrombozytopenie ausgeschlossen sind.
Diagnosekriterien für die Diagnose:
Beschwerden:
verstärkte Blutung aus Schleimhäuten;
Anamnese:
· Nasenbluten, Zahnfleischbluten;
· Menorrhagie, Metrorrhagie;
· Blutungen in der Sklera;
· Blutungen im Gehirn;
· Hämaturie;
· Blutungen aus dem Magen-Darm-Trakt (blutiges Erbrechen, Melena);
· hämorrhagische Hautausschläge in Form von Petechien und Ekchymosen.
Körperliche Untersuchung:
Allgemeine Inspektion:
Charakter des kutanen hämorrhagischen Syndroms:
· Lage und Größe der Petechien und Blutergüsse;
· Vorhandensein von Blutungen auf der Mundschleimhaut und den Bindehäuten;
· Blut fließt in den Rachenraum;
· Anomalien der Struktur des Gesichts (dreieckiges Gesicht, kleine Augen, Epikanthus, kleine Gesichtszüge) und der Gliedmaßen (Anomalien des 1. Fingers, des Sechsfingers, Syndaktylie, Klinodaktylie);
Laborforschung:
· Blutbild mit manueller Berechnung der Leukozytenformel und der Thrombozytenmorphologie – im Hämogramm Es wird eine isolierte Thrombozytopenie festgestellt – eine Abnahme der Blutplättchen um weniger als 100 x 10 9 / l ohne Veränderungen der Leukozyten- und Erythrogrammparameter. In einigen Fällen können eine posthämorrhagische Anämie und Veränderungen im Leukogramm im Zusammenhang mit einer begleitenden Infektionskrankheit oder Allergie festgestellt werden;
Nein.
Diagnosealgorithmus auf ambulanter Ebene:

Diagnostik (Krankenhaus)
DIAGNOSE AUF STATIONÄRER EBENE
Diagnosekriterien:
Beschwerden: siehe ambulante Ebene.
Anamnese:
Dauer und Art der Blutung;
· Impfung (insbesondere Kombinationsimpfung gegen Masern, Mumps und Röteln) 2-3 Wochen vor der Entwicklung eines hämorrhagischen Syndroms;
· übertragen (Atemwegsvirus, Röteln, infektiöse Mononukleose) 2-3 Wochen vor der Entwicklung des hämorrhagischen Syndroms;
· Einnahme von Medikamenten (insbesondere Heparin) in den letzten 2-3 Wochen;
Vorhandensein von Knochenschmerzen und Gewichtsverlust;
Körperliche Untersuchung: siehe ambulante Ebene .
Laborforschung:
· UAC mit manueller Berechnung der Leukozytenformel und der Thrombozytenmorphologie – das Hämogramm zeigt eine isolierte Thrombozytopenie – eine Abnahme der Thrombozyten um weniger als 100x109/l ohne Veränderung der Leukozyten- und Erythrogrammwerte. In einigen Fällen können eine posthämorrhagische Anämie und Veränderungen im Leukogramm im Zusammenhang mit einer begleitenden Infektionskrankheit oder Allergie festgestellt werden;
Instrumentalstudium: Nein.
Diagnosealgorithmus auf stationärer Ebene: Nein.
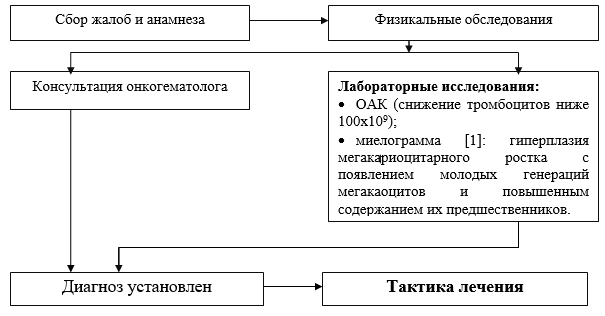
Liste der auf Krankenhausebene durchgeführten Basisdiagnostikmaßnahmen:
· CBC (Zählung von Blutplättchen und Retikulozyten in einem Abstrich);
· Blutgruppe und Rh-Faktor;
· biochemischer Bluttest (Protein, Albumin, ALT, ACaT, Bilirubin, Kreatinin, Harnstoff, Dextrose);
· Myelogramm: Hyperplasie der Megakaryozytenlinie mit dem Auftreten junger Megakaozytengenerationen und einem erhöhten Gehalt ihrer Vorläufer;
· Blutungsdauer nach Sucharew;
· OAM;
· ELISA für Marker der Virushepatitis (HbsAg);
· ELISA für Marker der Virushepatitis HCV;
· ELISA für HIV-Marker.
Liste zusätzlicher diagnostischer Untersuchungen auf Krankenhausebene:
· biochemische Analyse: GGTP, Elektrolyte;
· Koagulogramm;
· ELISA für antithrombotische Antikörper;
Immunphänotypisierung peripherer Blutzellen;
· Immunogramm;
· Antiphospholipid-Antikörper;
· PCR für Virusinfektionen (Virushepatitis, Cytomegalievirus, Herpes-simplex-Virus, Epstein-Barr-Virus, Varizellen-/Zoster-Virus);
· Echokardiographie;
· Ultraschall der Bauchorgane (Leber, Milz, Bauchspeicheldrüse, Gallenblase, Lymphknoten, Nieren), Mediastinum, Retroperitoneum und Becken – um Blutungen in den inneren Organen auszuschließen;
· Computertomographie des Gehirns: wird durchgeführt, wenn der Verdacht auf eine intrakranielle Blutung besteht – Kopfschmerzen, Erbrechen, Paresen, Bewusstseinsstörungen; um einen Schlaganfall auszuschließen;
· Ultraschall von OBP.
Differenzialdiagnose
| Diagnose | Begründung für die Differentialdiagnose | Umfrage | Ausschlusskriterien für die Diagnose |
| TAR-Syndrom | Gekennzeichnet durch Pathologie von Megakaryozyten und Blutplättchen mit ihrer Hypoplasie und Funktionsstörung, die zu Blutungen führt | Erhebung von Beschwerden und Anamnese, Methode der körperlichen Untersuchung. | Gekennzeichnet durch das Fehlen von Radiusknochen, angeborene Pathologie von Megakaryozyten und Blutplättchen mit ihrer Hypoplasie und Funktionsstörung, die zu Blutungen führt. Kinderkrankheiten gehen am häufigsten mit angeborenen Organanomalien (häufig Herzfehlern) einher. |
| Aplastische Anämie | In Blutausstrichen ist die isolierte Thrombozytopenie häufig so ausgeprägt, dass einzelne Blutplättchen nachgewiesen werden. | Blutbild mit Zählung der Leukozytenformel, Retikulozyten. Myelogramm, Trepanbiopsie. | Knochenmarkaspirat ist arm an Kernelementen. Der Gesamtanteil zellulärer Elemente wird reduziert. In histologischen Präparaten von Trepanbiopsien der Beckenknochen schließt eine Knochenmarkaplasie mit Ersatz von Fettgewebe eine ITP aus. Der Eisengehalt ist normal oder erhöht. |
| Myelodysplastisches Syndrom | Hämorrhagisches Syndrom | Blutbild (mit Leukozytenzahl, Retikulozytenzahl). Myelogramm, Trepanbiopsie. | MDS ist durch Anzeichen einer Dyspoese, übermäßige Blasten im Knochenmark und Chromosomenaberrationen gekennzeichnet, was eine ITP ausschließt |
| Hämatoblastosen | Panzytopenie, hämorrhagisches Syndrom | Blutbild (mit Leukozytenzahl, Retikulozytenzahl). Myelogramm. | Die Ergebnisse der Durchflusszytometrie sowie der immunhistochemischen und histologischen Untersuchung des Knochenmarks schließen eine ITP aus. |
| Paroxysmale nächtliche Hämoglobinurie | Hämorrhagisches Syndrom |
UAC; Blutchemie; Koagulogramm; OAM; IFT auf PNG. |
PNH ist durch Hämosiderinurie, Hämoglobinurie, erhöhte Bilirubin- und LDH-Spiegel sowie eine Abnahme oder Abwesenheit von Haptoglobin gekennzeichnet. Blutungen werden selten beobachtet, Hyperkoagulation (Aktivierung von Aggregationsinduktoren) ist typisch. Ausgeschlossen, wenn auf Grundlage der IFT-Ergebnisse kein PNH-Klon vorliegt. |
| Megaloblastenanämie. | Thrombozytopenie |
CBC + periphere Blutmorphologie; Myelogramm; Biochemischer Bluttest (Cyanocobalamin- und Folsäurespiegel). |
Indirekte Anzeichen, die für eine megaloblastäre Anämie charakteristisch sind, sind ein Anstieg des durchschnittlichen Hämoglobingehalts in Erythrozyten, ein Anstieg des durchschnittlichen Erythrozytenvolumens und der megaloblastäre Typ der Hämatopoese laut Myelogramm. Im Gegensatz zur ITP liegt bei der Megaloblastenanämie trotz Thrombozytopenie kein hämorrhagisches Syndrom vor. |
| Thrombotische thrombozytopenische Purpura. | Hämorrhagisches Syndrom |
UAC; Ultraschall von OBP; Beurteilung des neurologischen Status; Röntgen der Gelenke. |
Sie wird aufgrund neurologischer Symptome, der Bildung mehrerer Blutgerinnsel, eines Gelenksyndroms und häufig einer Vergrößerung von Leber und Milz ausgeschlossen. |
Behandlung im Ausland
Lassen Sie sich in Korea, Israel, Deutschland und den USA behandeln
Lassen Sie sich zum Thema Medizintourismus beraten
Behandlung
Medikamente (Wirkstoffe), die bei der Behandlung eingesetzt werden
| Blutstillender Schwamm |
| Azithromycin |
| Alemtuzumab |
| Amoxicillin |
| Aciclovir |
| Dexamethason |
| Immunglobulin G vom Menschen normal (Immunglobulin G vom Menschen normal) |
| Captopril |
| Clavulansäure |
| Kolekaltsiferol |
| Thrombozytenkonzentrat (CT) |
| Mycophenolsäure (Mycophenolatmofetil) |
| Omeprazol |
| Pankreatin |
| Paracetamol |
| Piperacillin |
| Prednisolon |
| Rituximab |
| Tazobactam |
| Tranexamsäure |
| Thrombinum |
| Fluconazol |
| Ceftazidim |
| Cyclosporin |
| Cyclophosphamid |
| Eltrombopag |
| Etamsylat |
Behandlung (Ambulanz)
Ambulante Behandlung
Behandlungstaktiken: Nein.
− Nichtmedikamentöse Behandlung: Nein.
− Medikamentöse Behandlung: Nein.
Aktionsalgorithmus in Notsituationen: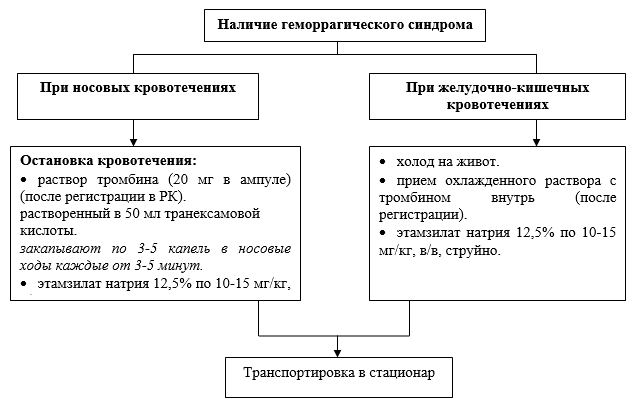
· Konsultation eines Onkohämatologen – bei Verdacht auf Hämatoblastose;
· Konsultation eines Gynäkologen – bei Metrorrhagie, Menorrhagie;
Behandlung (Krankenwagen)
DIAGNOSE UND BEHANDLUNG IN DER NOTFALLVERSORGUNG
Diagnostische Maßnahmen:
· Sammlung von Beschwerden und Krankengeschichte;
· körperliche Untersuchung.
Medikamentöse Behandlung:
· symptomatische Therapie ,
gemäß IMCI – WHO-Richtlinien für die Behandlung der häufigsten Krankheiten in Krankenhäusern der Primärstufe, angepasst an die Bedingungen der Republik Kasachstan.
Behandlung (stationär)
STATIONÄRE BEHANDLUNG
Behandlungstaktiken:
Bei Immunthrombozytopenie beginnt die Behandlungstaktik mit der Verschreibung eines Hormonpräparats (Prednisolon). Bei positivem Ansprechen auf die Behandlung steigt die Thrombozytenzahl (normalerweise an den Tagen 7–10) und bleibt auch nach Absetzen des Arzneimittels auf einem hohen Niveau. Tritt keine Remission ein, wird eine Immuntherapie verschrieben – intravenöses Immunglobulin. Kann der Patient mit einer medikamentösen Therapie nicht innerhalb von 6 Monaten in eine Remission gebracht werden, wird eine Splenektomie empfohlen. In schweren Fällen der Erkrankung kann die Splenektomie zu einem früheren Zeitpunkt durchgeführt werden.
Um eine Entscheidung über die Behandlungstaktik zu treffen, hat eine internationale Expertengruppe eine Blutungsskala und Empfehlungen für das Vorgehen entwickelt
zur Therapie:
| Blutung/Lebensqualität | Therapeutischer Ansatz |
|
Abschluss 1. Leichte Blutung<100 петехий и/или < 5 мелких синяков (<3 см в диаметре); отсутствие кровоточивости слизистых |
Überwachung |
|
Abschluss 2. Leichte Blutung. Mehrere Petechien > 100; und/oder >5 große blaue Flecken (>3 cm Durchmesser); kein Bluten der Schleimhäute |
Beobachtung oder bei einigen Patienten membranstabilisierende Therapie |
|
Abschluss 3. Mäßige Blutung. Das Vorhandensein von Schleimhautblutungen, ein „gefährlicher“ Lebensstil |
Rücksprache mit einem Hämatologen |
|
Grad 4. Blutende Schleimhäute oder Verdacht auf innere Blutung |
Behandlung aller Patienten im Krankenhausumfeld |
Nichtmedikamentöse Behandlung:
Modus: II.III;
Diät: № 11.
Medikamentöse Behandlung
Behandlung je nach Schweregrad:
Verwenden Sie die Standarddosis Prednisolon maximal 14 Tage lang/erhöhte Dosis 4 Tage lang
Erstbehandlung bei ITP:
| Drogen | Dosis | Dauer der Therapie |
UD, Verknüpfung |
| Prednisolon | 0,25 mg/kg | 21 Tage | Klasse A |
| 2 mg/kg | 14 Tage mit schrittweisem Rückzug | ||
| 60 mg/m² | 21 Tage | ||
| 4 mg/kg | 7 Tage mit schrittweisem Rückzug | ||
| 4 mg/kg | 4 Tage | ||
| Methylprednisolon | 30 oder 50 mg/kg | 7 Tage | Klasse A |
| 20-30 mg/kg | 2 - 7 Tage | ||
| 30 mg/kg | 3 Tage | ||
| IVIG | 0,8-1 g/kg | 1-2 Tage | Klasse A |
| 0,25 g/kg | Einmal | ||
| 0,4 g/kg | 5 Tage | ||
| Anti-D | 25µg/kg | 2 Tage | Klasse A |
| 50-60 µg/kg | Einmal | ||
| 75mcg/kg | Einmal | ||
| Dexamethason | 20 - 40 mg/kg/Tag | für 4 aufeinanderfolgende Tage (jeden Monat, 6 Zyklen) | Klasse A |
Anhaltende und chronische ITP:
· Glukokortikoid-Therapieschemata: hohe Dosen Methylprednisolon i.v. 30 mg/kg x 3 Tage, dann 20 mg/kg x 4 Tage;
· IVIT kann auch bei CITP, vor chirurgischen Eingriffen, Zahnextraktion/bei Verletzungen eingesetzt werden. Die Behandlungsschemata für die Verwendung von IVIT bei cITP sind identisch mit denen bei neu aufgetretener ITP.
· Die empfohlene IVIT-Dosis beträgt 0,8-1,0 g/kg Körpergewicht, gefolgt von einer wiederholten Verabreichung innerhalb von 48 Stunden, wenn nach der ersten Verabreichung der Thrombozytenspiegel nicht höher als 20 x 109/l ist.
Medikamentöse Zweitlinientherapie:
Rituximab(UD-B):
· Einzeldosis: 375 mg/m2/Woche, Kurdauer: 4 Wochen (insgesamt 4 Injektionen);
Hinweise:
· Nichtansprechen auf hohe Dosen Dexamethason;
· wenn Kontraindikationen für eine Splenektomie bestehen;
· wiederkehrender und refraktärer Verlauf der ITP.
Cyclosporin A:
· 2,5 - 3 mg/kg/Tag. In Kombination mit Prednisolon (UD-B)
Cyclophosphamid: 200 mg/m2 1 Mal pro Tag;
Hinweise:
· bei Patienten, die gegen eine Hormontherapie resistent sind und/oder nach einer Splenektomie;
· sekundäres ITP.
Mycophenolatmofetin: 20-40 mg/kg, Kursdauer 30 Tage.
Hinweise:
· einige Patienten mit antiproliferativen und immunsuppressiven Zwecken.
Arzneimitteltherapie der dritten Linie:
TPO-Rezeptoragonisten(UD-A):
· Eltrombopag 25–75 mg oral 1–10 mg/kg/Woche.
Alemtuzumab*:
· Alternative Therapie für cITP und refraktäre ITP.ACHTUNG! wird vor dem Hintergrund einer begleitenden Therapie (antibakteriell, antimykotisch, antiviral) eingesetzt.
Liste der wichtigsten Medikamente:
| INN der Droge | Freigabe Formular |
UD, Verknüpfung |
| Immunsuppressive Medikamente | ||
| Dexamethason |
Tabletten 0,5 mg Lösung 4 mg/2 ml |
UD B |
| Prednisolon | 5 mg Tabletten | UD A |
| zur intravenösen Verabreichung 10 % 2 g/20 ml | UD A | |
| Immunglobulin menschliches Ig G | zur intravenösen Verabreichung 10 % 5 g/50 ml | UD A |
| Cyclophosphamid | Pulver zur Herstellung einer Lösung zur intravenösen Verabreichung 500 mg | UD S |
| Mycophenolatmofetil | Kapseln 250 und 500 mg | UD S |
| Rituximab |
Flaschen 10 ml/100 mg Flaschen 50 ml/500 mg |
UD B |
| Cyclosporin A | Kapseln 25 mg, 50 mg, 100 mg | UD B |
| Eltrombopag | Tabletten 31,9 mg und 63,8 mg | UD A |
| Alemtuzimab (nach Registrierung in der Republik Kasachstan) | Infusionslösung 1 ml | UD A |
| Antimykotische Medikamente(nach Angaben) | ||
| Fluconazol | Lösung zur intravenösen Injektion, 50 ml, 2 mg/ml, Kapseln 150 mg | UD B |
| Antimikrobielle Mittel Wird zur Vorbeugung der Entwicklung eitrig-septischer Komplikationen sowie zur Feststellung der Empfindlichkeit gegenüber Antibiotika eingesetzt | ||
|
Azithromycin oder |
Tablette/Kapsel, 500 mg, lyophilisiertes Pulver zur Herstellung einer Lösung zur intravenösen Infusion, 500 mg; | UD B |
|
Piperacillin/Tazobactam oder |
Pulver zur Herstellung einer Injektionslösung zur intravenösen Verabreichung 4,5 g | UD B |
|
Ceftazidim oder |
Pulver zur Herstellung einer Injektionslösung zur intravenösen Verabreichung 1000 mg | UD B |
| Amoxocillin + Clavulansäure |
Filmtablette, 500 mg/125 mg, Pulver zur Herstellung einer Suspension zum Einnehmen 135 mg/5 ml, Pulver zur Herstellung einer Lösung zur intravenösen und intramuskulären Verabreichung 600 mg. |
UD B |
| Virostatikum ( je nach Indikation, bei Infektion) | ||
| Aciclovir | Creme zur äußerlichen Anwendung 5 % -5,0, Tablette 200 mg, Pulver zur Herstellung einer Infusionslösung 250 mg; | UD S |
| Arzneimittel, die das Blutgerinnungssystem beeinflussen | ||
| Fibrinogen+Thrombin | blutstillender Schwamm, Größe 7*5*1, 8*3; | UD B |
Liste zusätzlicher Medikamente:
| INN der Droge |
Verabreichungsweg |
UD, Verknüpfung |
| Omeprazol (Vorbeugung einer Anti-Geschwür-Therapie) | zur oralen Verabreichung 20 mg | UD B |
| Pankreatin (bei Gastritis, verbessert den Verdauungsprozess durch Hormontherapie) | 10000 IE | UD B |
| Captopril (gegen erhöhten Blutdruck) | Tablette zur oralen Verabreichung 12,5 mg | UD B |
| Paracetamol (fiebersenkend) | Tablette zur oralen Verabreichung 200 mg | UD B |
| Natriumethamsylat (gegen Blutungen) |
zur oralen Verabreichung zur intravenösen Verabreichung 2 ml |
UD B |
| Colecalciferol (gegen Hypokalzämie) | 500 mg Tabletten | UD B |
Einsatz von Thrombozytenkonzentrat-Transfusionen:
Hinweise:
· Vorliegen lebensbedrohlicher Blutungen.
Transfusionen von Thrombozytenkonzentrat sollten immer eine spezifische Therapie bei ITP (IVIG und/oder Glukokortikoide) ergänzen und nicht als Monotherapie eingesetzt werden. Wenn die Blutung bei ITP so stark ist, dass eine Transfusion von Thrombozytenkonzentrat erforderlich ist, werden Split-Transfusionen empfohlen – alle 6–8 Stunden. In besonders schweren Fällen werden „hyperfraktionierte“ Transfusionen mit kleinen Dosen Thrombozytenkonzentrat eingesetzt: 1-2 Dosen (0,7-1,4x10 11) alle zwei Stunden. Als zusätzliche blutstillende Therapie werden Etamsylat und Antifibrinolytika eingesetzt.
ACHTUNG! Bei Nierenblutungen ist die Gabe von Fibrinolysehemmern kontraindiziert.
Operativer Eingriff:
Splenektomie(UD-B)
Indikationen für eine Intervention:
· wiederkehrender, schwerer Krankheitsverlauf über mehr als 6 Monate;
· Patienten über 6 Jahre nach vorheriger Impfung mit Haemophilus influenzae Typ b + S.pneumoniae + N.Meningitidis.
Kontraindikationen für einen Eingriff:
· Kinder unter 6 Jahren;
· primäres ITP.
Andere Behandlungen: Nein.
Zusätzliche hämostatische Therapie:
· Natriumethamsylat 12,5 % in einer Dosis von 10-15 mg/kg;
· para-Aminobenzoesäure – Tranexamsäure: über 12 Jahre in einer Dosis von 20–25 mg/kg.
Hinweise zur Konsultation von Spezialisten:
· Konsultation eines Spezialisten für Infektionskrankheiten – bei Verdacht auf einen infektiösen Prozess;
· Konsultation eines Endokrinologen – wenn sich während der Behandlung endokrine Störungen entwickeln;
· Rücksprache mit einem Geburtshelfer-Gynäkologen – während der Schwangerschaft, Metrorrhagie, Menorrhagie, bei der Verschreibung kombinierter oraler Kontrazeptiva;
· Rücksprache mit anderen Fachärzten – je nach Indikation.
Indikationen zur Verlegung auf die Intensivstation:
· Abwesenheit/Bewusstseinsstörung (Glasgow-Skala); Antrag Nr. 1
akutes Herz-Kreislauf-Versagen (Herzfrequenz unter 60 oder mehr als 200 pro Minute);
· akutes Atemversagen (Atembeschwerden 2–3 Grad, Atemfrequenz mehr als 50, verringerte Sättigung weniger als 88 %, Notwendigkeit einer mechanischen Beatmung);
Akute Durchblutungsstörungen (Schockzustände);
· Systolischer Blutdruck, weniger als 60/mehr als 180 (erfordert die ständige Verabreichung vasoaktiver Medikamente);
· kritische Stoffwechselstörungen (Elektrolyt, Wasser, Eiweiß, Säure-Basen-Haushalt, Ketoazidose);
· intensive Beobachtung und intensive Pharmakotherapie, die eine ständige Überwachung der Vitalfunktionen erfordern;
· Verletzung der Blutgerinnungs- und Antikoagulationssysteme.
Indikatoren für die Wirksamkeit der Behandlung:
· 4 Wochen nach Beginn der Behandlung ein Anstieg der Blutplättchen über 100 x 10 9 / l (75 % der Patienten mit ITP).
· nach Entfernung der Milz – ein Anstieg der Blutplättchen im peripheren Blut.
Weitere Verwaltung
Laborforschung:
· Ein Blutbild mit Bestimmung der Thrombozytenzahl und manueller Auszählung der Leukozytenformel (obligatorisch) wird im ersten Beobachtungsjahr einmal monatlich durchgeführt. Darüber hinaus abhängig vom klinischen Zustand und der Stabilität des hämatologischen Bildes;
· Bei Bedarf wird eine dynamische biochemische Blutanalyse durchgeführt.
· Serologische Tests auf Marker für HIV, Hepatitis B und C, durchgeführt 3 Monate nach der Entlassung aus dem Krankenhaus und 3 Monate nach jeder Transfusion von Blutprodukten.
Voraussetzung für die Überstellung des Patienten an den Wohnort:
· Der Kinderarzt (Kinderhämatologe) am Wohnort orientiert sich an den Empfehlungen der Krankenhausspezialisten.
· Die Häufigkeit der Untersuchung eines Patienten mit ITP beträgt in den ersten 3 Behandlungsmonaten einmal alle 2–4 Wochen, danach je nach klinischem Zustand und hämatologischer Dynamik, mindestens jedoch alle 2 Monate.
Instrumentalstudien bei klinischer Indikation durchgeführt werden.
Krankenhausaufenthalt
Hinweise auf einen geplanten Krankenhausaufenthalt:
Indikationen für einen Notfall-Krankenhausaufenthalt:
Abnahme des Blutplättchenspiegels im Blutbild<50х10 9 /л.
· Vorliegen eines hämorrhagischen Syndroms (Blutungen aus den Schleimhäuten des Nasopharynx, der Mundhöhle, Magen-Darm-Blutungen, Uterusblutungen).
Information
Quellen und Literatur
- Protokolle der Sitzungen der Gemeinsamen Kommission für die Qualität medizinischer Dienstleistungen des Gesundheitsministeriums der Republik Kasachstan, 2016
- 1) Pädiatrische Hämatologie, 2015. Herausgegeben von A.G. Rumyantsev, A.A. Maschan, E.V. Zhukovskaya. Moskau. Verlagsgruppe „GEOTAR-Media“ 2015 C – 656, C-251, Tabelle 6. 2) Evidenzbasierte Praxisleitlinie der American Society of Hematology 2011 für Immunthrombozytopenie Cindy Neunert, Wendy Lim, Mark Crowther, Alan Cohen, Lawrence Solberg, Jr. und Mark A. Crowther2011; 16:4198-4204 3) Standardisierung von ITP, September 2006 IMBACH. 4) Bereitstellung von Notfallversorgung, 2005. Aktionsalgorithmus in Notfallsituationen: gemäß IMCI – WHO-Richtlinien für die Behandlung der häufigsten Krankheiten in Primärkrankenhäusern, angepasst an die Bedingungen der Republik Kasachstan (WHO 2012). 5) ESH. The Handbook „Immune thrombocytopenia“ 2011. 6) Tarantino & Buchanan, Hematol Oncol Clin North Am, 2004, 18:1301-1314. 7) Richtlinien für die Verwaltung parenteraler Ernährung Kanada 2010. 8) SIGN 104. Antibiotikaprophylaxe in der Chirurgie.2014.
Information
Im Protokoll verwendete Abkürzungen
| AG | arterieller Hypertonie; |
| HÖLLE | arterieller Druck; |
| ALaT | Alanin-Aminotransferase |
| ASa T | Aspartat-Aminotransferase |
| IV | intravenös |
| Ich bin | intramuskulär |
| VVID | intravenöse hochdosierte Immunglobulintherapie |
| HIV | AIDS-Virus; |
| GGTP | Gammaglutamyl-Transpeptidase; |
| IMCI | Integriertes Management von Kinderkrankheiten |
| mechanische Lüftung | künstliche Beatmung |
| USW | Immunthrombozytopenie |
| ELISA | verknüpfter Immunosorbens-Assay; |
| IFT | Immunphänotypisierung; |
| CT | CT-Scan; |
| KSH | Säure-Base-Zustand |
| LDH | Laktatdehydrogenase; |
| Gesundheitseinrichtungen | medizinische Einrichtung |
| MDS | myelodysplastisches Syndrom; |
| MICH | Internationale Einheiten |
| Geldmarktfonds | Mycophenolatmofetin |
| MRT | Magnetresonanztomographie |
| UAC | allgemeine Blutanalyse |
| OAM | allgemeine Urinanalyse; |
|
AML PNG |
akute myeloblastische Leukämie; paroxysmale nächtliche Hämoglobinurie; |
| ONMK | akuter zerebrovaskulärer Unfall |
| PCR | Polymerase Kettenreaktion; |
| ESR | - Blutsenkungsgeschwindigkeit; |
| HSCT | hämatopoetische Stammzelltransplantation |
| USDG | Doppler-Ultraschall |
| FGDS | Fibro-Gastro-Duadenoskopie |
| hITP | chronische Immunthrombozytopenie |
| CMV | Cytomegalovirus |
| BH | Atmungsrate; |
| Pulsschlag | Pulsschlag; |
| EKG | Elektrokardiographie; |
| EchoCG | Echokardiographie; |
| Ich G | Immunoglobulin |
Liste der Protokollentwickler mit Qualifikationsinformationen:
1) Omarova Gulnara Erbosynovna – pädiatrische Hämatologin/Onkologin, Zweigstelle der Unternehmensstiftung „UMC“, „Nationales Wissenschaftszentrum für Mutterschaft und Kindheit“, Astana.
2) Tastanbekova Venera Bulatovna – pädiatrische Hämatologin/Onkologin, Zweigstelle der Unternehmensstiftung „UMC“, „Nationales Wissenschaftszentrum für Mutterschaft und Kindheit“, Astana.
3) Umirbekova Balzhan Bolatovna – pädiatrische Hämatologin/Onkologin, Zweigstelle der Unternehmensstiftung „UMC“, „Nationales Wissenschaftszentrum für Mutterschaft und Kindheit“, Astana.
4) Omarovna Kulyan Omarovna – Doktor der medizinischen Wissenschaften, Professorin, Nationales Zentrum für Pädiatrie und Kinderchirurgie, Almaty.
5) Manzhuova Lyazzat Nurpapaevna – Kandidatin der medizinischen Wissenschaften, Leiterin der Abteilung für Onkologie Nr. 1, Nationales Zentrum für Pädiatrie und Kinderchirurgie, Almaty.
6) Mira Maratovna Kalieva – Kandidatin der medizinischen Wissenschaften, außerordentliche Professorin der nach ihr benannten Abteilung für klinische Pharmakologie und Pharmakotherapie der KazNMU. S. Asfendiyarova.
Hinweis auf keine Konflikte: Nein.
Liste der Gutachter: Kemaykin Vadim Matveevich – Hämatologe der höchsten Qualifikationskategorie, Kandidat der medizinischen Wissenschaften, leitender freiberuflicher Hämatologe, Onkohämatologe des Ministeriums für Gesundheit und soziale Entwicklung der Republik Kasachstan.
Anhang 1

Angehängte Dokumente
Aufmerksamkeit!
- Durch Selbstmedikation können Sie Ihrer Gesundheit irreparablen Schaden zufügen.
- Die auf der MedElement-Website und in den mobilen Anwendungen „MedElement“, „Lekar Pro“, „Dariger Pro“, „Diseases: Therapist's Guide“ veröffentlichten Informationen können und sollen eine persönliche Konsultation mit einem Arzt nicht ersetzen. Wenden Sie sich unbedingt an eine medizinische Einrichtung, wenn Sie Krankheiten oder Symptome haben, die Sie beunruhigen.
- Die Wahl der Medikamente und deren Dosierung müssen mit einem Facharzt besprochen werden. Nur ein Arzt kann unter Berücksichtigung der Erkrankung und des Zustands des Körpers des Patienten das richtige Arzneimittel und dessen Dosierung verschreiben.
- Die MedElement-Website und die mobilen Anwendungen „MedElement“, „Lekar Pro“, „Dariger Pro“, „Krankheiten: Therapeutenverzeichnis“ sind ausschließlich Informations- und Referenzressourcen. Die auf dieser Website veröffentlichten Informationen dürfen nicht dazu verwendet werden, ärztliche Anordnungen unbefugt zu ändern.
- Die Herausgeber von MedElement haften nicht für Personen- oder Sachschäden, die durch die Nutzung dieser Website entstehen.
ICD-10-CODE
FANCONI-ANÄMIE
DIAMOND-BLACKFAN-ANÄMIE ICD-10-CODE
ICD-10-CODE
D61. Andere aplastische Anämien. Arten von AA:
Angeboren [Fanconi-Anämie (FA), Diamond-Blackfan-Anämie (DBA), Dyskeratosis congenita, Shwachman-Diamond-Oski-Anämie, Amegakaryozyten-Thrombozytopenie];
Erworben (idiopathisch, verursacht durch Viren, Medikamente oder Chemikalien).
AA kommt mit einer Häufigkeit von 1–2 Fällen pro 1.000.000 Einwohner pro Jahr vor und gilt als seltene Blutkrankheit. Erworbene AA entwickelt sich mit einer Häufigkeit von 0,2–0,6 Fällen pro 100.000 Kindern pro Jahr. Die durchschnittliche jährliche Inzidenzrate von AA bei Kindern betrug von 1979 bis 1992 in der Republik Belarus 0,43 ± 0,04 pro 100.000 Kinder. Es gab keine Unterschiede in der Inzidenz von AA bei Kindern vor und nach der Katastrophe von Tschernobyl.
DBA wird unter vielen Namen beschrieben; partielle Erythrozytenaplasie, angeborene hypoplastische Anämie, echte Erythrozytenanämie, primäre Erythrozytenerkrankung, Erythrogenesis imperfecta. Die Krankheit ist selten, L.K. Diamond et al. in den 60ern 20. Jahrhundert beschrieb nur 30 Fälle dieser Krankheit; bisher wurden mehr als 400 Fälle beschrieben.
Lange Zeit ging man davon aus, dass die Inzidenz von DBA bei 1 Fall pro 1.000.000 lebenden Neugeborenen liegt. Im Jahr 1992 berichtete L. Wranne über eine höhere Inzidenz von 10 Fällen pro 1.000.000 Geburten. Die Inzidenzrate von DBA beträgt laut französischen und englischen Registern 5–7 Fälle pro 1.000.000 lebende Neugeborene. Das Geschlechterverhältnis ist nahezu gleich. Mehr als 75 % der DBA-Fälle sind sporadisch; 25 % sind familiärer Natur und in einigen Familien sind mehrere Patienten registriert. Das Patientenregister mit DBA in den USA und Kanada umfasst 264 Patienten im Alter von 10 Monaten bis 44 Jahren.
D61.0. Konstitutionelle aplastische Anämie.
FA ist eine seltene autosomal-rezessive Erkrankung, die durch mehrere angeborene körperliche Anomalien, fortschreitendes Knochenmarkversagen und eine Veranlagung zur Entwicklung bösartiger Erkrankungen gekennzeichnet ist. Die Inzidenz von Vorhofflimmern beträgt 1 Fall pro 360.000–3.000.000 Einwohner. Die Krankheit kommt bei allen Nationalitäten und ethnischen Gruppen vor. Das Mindestalter für die Manifestation klinischer Symptome ist die Neugeborenenperiode, das Höchstalter liegt bei 48 Jahren. Das Patientenregister mit Vorhofflimmern des Forschungsinstituts für pädiatrische Hämatologie des Gesundheitsministeriums der Russischen Föderation erfasste die Daten von 69 Patienten. Das durchschnittliche Manifestationsalter der Krankheit beträgt 7 Jahre (2,5–12,5 Jahre). Es wurden 5 familiäre Fälle identifiziert.
HÄMORRHAGISCHE ERKRANKUNGEN Purpura und andere hämorrhagische Erkrankungen
D69.3. Idiopathische thrombozytopenische Purpura.
Nach Ansicht vieler Hämatologen ist die idiopathische thrombozytopenische Purpura (ITP) eine häufige hämorrhagische Erkrankung. Die einzige Studie in unserem Land zeigt jedoch, dass die Inzidenzrate von ITP in der Region Tscheljabinsk 3,82 ± 1,38 Fälle pro 100.000 Kinder pro Jahr beträgt und keinen steigenden Trend aufweist.
In Russland wurde die Internationale Klassifikation der Krankheiten, 10. Revision (ICD-10), als einziges normatives Dokument zur Erfassung von Morbidität, Gründen für Besuche der Bevölkerung in medizinischen Einrichtungen aller Abteilungen und Todesursachen übernommen.
ICD-10 wurde 1999 auf Anordnung des russischen Gesundheitsministeriums vom 27. Mai 1997 in der Gesundheitspraxis in der gesamten Russischen Föderation eingeführt. Nr. 170
Die Veröffentlichung einer neuen Revision (ICD-11) ist von der WHO für 2017-2018 geplant.
Mit Änderungen und Ergänzungen der WHO.
Bearbeitung und Übersetzung von Änderungen © mkb-10.com
Thrombozytopenie – Beschreibung, Ursachen, Symptome (Anzeichen), Diagnose, Behandlung.
Kurzbeschreibung
Thrombozytopenie ist eine niedrige Thrombozytenzahl im peripheren Blut und die häufigste Ursache für Blutungen. Wenn die Thrombozytenzahl auf weniger als 100 x 109/l sinkt, verlängert sich die Blutungszeit. In den meisten Fällen treten Petechien oder Purpura auf, wenn die Thrombozytenzahl auf 20–50 ´ 109/l absinkt. Bei einer Thrombozytopenie unter 10 ´ 109/L kommt es zu schweren spontanen Blutungen (z. B. Magen-Darm-Blutungen) oder zu hämorrhagischen Schlaganfällen.
Ursachen
Thrombozytopenie kann als Manifestation einer Arzneimittelallergie (allergische Thrombozytopenie), verursacht durch die Produktion von Thrombozytenaggregationshemmern (autoimmune Thrombozytopenie), verursacht durch Infektionen, Intoxikationen, Thyreotoxikose (symptomatisch) auftreten.
Bei Neugeborenen kann eine Thrombozytopenie durch das Eindringen von Autoantikörpern einer erkrankten Mutter durch die Plazenta verursacht werden (transimmune Thrombozytopenie).
Pathologie der Thrombozytopoese Die Reifung von Megakaryozyten wird durch Thiaziddiuretika und andere Medikamente, insbesondere solche, die in der Chemotherapie verwendet werden, selektiv unterdrückt, Ethanol. Eine besondere Ursache der Thrombozytopenie ist eine ineffektive Thrombopoese, die mit dem megaloblastischen Typ der Hämatopoese verbunden ist (tritt bei einem Mangel an Vitamin B 12 auf und). Folsäure sowie bei myelodysplastischen und präleukämischen Syndromen). Im Knochenmark werden morphologisch und funktionell abnormale (megaloblastische oder dysplastische) Megakaryozyten identifiziert, die zu einer Ansammlung defekter Blutplättchen führen, die im Knochenmark zerstört werden. Amegakaryozyten-Thrombozytopenie ist eine seltene Ursache für Thrombozytopenie, die durch einen angeborenen Mangel an Megakaryozyten-Kolonie verursacht wird. Einheiten bilden.
Anomalien in der Bildung des Blutplättchenpools treten auf, wenn Blutplättchen aus dem Blutkreislauf eliminiert werden; die häufigste Ursache ist die Ablagerung in der Milz. Unter normalen Bedingungen enthält die Milz ein Drittel des Blutplättchenpools. Die Entwicklung einer Splenomegalie geht mit der Ablagerung einher einer größeren Anzahl von Zellen mit deren Ausschluss aus dem Blutstillungssystem. Bei einer sehr großen Milzgröße ist es möglich, 90 % des gesamten Blutplättchenpools abzulagern, die restlichen 10 % haben eine normale Zirkulationsdauer im peripheren Blutkreislauf.
Eine verstärkte Zerstörung von Blutplättchen in der Peripherie ist die häufigste Form der Thrombozytopenie; Solche Erkrankungen sind durch eine verkürzte Lebensdauer der Blutplättchen und eine erhöhte Anzahl von Megakaryozyten im Knochenmark gekennzeichnet. Diese Störungen werden als immun- oder nichtimmun-thrombozytopenische Purpura bezeichnet. Immun-thrombozytopenische Purpura. Die idiopathische thrombozytopenische Purpura (ITP) ist der Prototyp einer durch Immunmechanismen verursachten Thrombozytopenie (es gibt keine offensichtlichen äußeren Ursachen für die Zerstörung von Blutplättchen). Siehe Idiopathische thrombozytopenische Purpura. Andere autoimmune Thrombozytopenien, die durch die Synthese von Thrombozytenaggregationshemmern verursacht werden: Thrombozytopenie nach der Transfusion (verbunden mit der Exposition gegenüber Isoantikörpern), medikamenteninduzierte Thrombozytopenie (z. B. verursacht durch Chinidin), Thrombozytopenie verursacht durch Sepsis (die Inzidenz kann 70 erreichen). %), Thrombozytopenie in Kombination mit SLE und anderen Autoimmunerkrankungen. Die Behandlung zielt auf die Korrektur der zugrunde liegenden Pathologie ab. Es ist notwendig, die Einnahme aller potenziell gefährlichen Medikamente abzubrechen. Die GK-Therapie ist nicht immer wirksam. Transfundierte Blutplättchen unterliegen der gleichen beschleunigten Zerstörung. Nichtimmune thrombozytopenische Purpura. Infektionen (z. B. viral oder Malaria). Massive Transfusion von konserviertem Blut mit niedrigem Blutplättchengehalt. DIC. Prothetische Herzklappen. Thrombotische thrombozytopenische Purpura.
Thrombozytopenie (*188000, Â). Klinische Manifestationen: Makrothrombozytopenie, hämorrhagisches Syndrom, Rippenaplasie, Hydronephrose, rezidivierende Hämaturie. Labortests: Autoantikörper gegen Blutplättchen, verkürzte Lebensdauer der Blutplättchen, verlängerte Gerinnungszeit, normaler Tourniquet-Test, Defekte in der Plasmakomponente der Blutstillung.
May-Hegglin-Anomalie (Hegglin-Syndrom, Â). Makrothrombozytopenie, basophile Einschlüsse in Neutrophilen und Eosinophilen (Döhle-Körperchen).
Epstein-Syndrom (153650, Â). Makrothrombozytopenie in Kombination mit Allport-Syndrom.
Fechtner-Familiensyndrom (153640, Â). Makrothrombozytopenie, Einschlüsse in Leukozyten, Nephritis, Taubheit.
Angeborene Thrombozytopenie (600588, Deletion 11q23.3–qter, Â). Klinische Manifestationen: angeborene Dysmegakaryozyten-Thrombozytopenie, leichtes hämorrhagisches Syndrom. Laboruntersuchungen: 11q23.3-qter-Deletion, erhöhte Anzahl von Megakaryozyten, Riesengranula in peripheren Blutplättchen.
Zyklische Thrombozytopenie (188020, Â). Hämorrhagisches Syndrom, zyklische Neutropenie.
Thrombozytopenie Paris–Trousseau (188025, Deletion 11q23, Defekt des TCPT-Gens, Â). Klinische Manifestationen: hämorrhagisches Syndrom, Thrombozytopenie, Hypertelorismus, Ohranomalien, geistige Behinderung, Aortenisthmusstenose, Entwicklungsverzögerung in der Embryonalperiode, Hepatomegalie, Syndaktylie. Laboruntersuchungen: Riesengranula in Blutplättchen, Megakaryozytose, Mikromegakaryozyten.
TAR-Syndrom (aus: Thrombozytopenie – fehlender Radius – Thrombozytopenie und Fehlen des Radius, *270400, r). Angeborenes Fehlen des Radius in Kombination mit Thrombozytopenie (bei Kindern ausgeprägt, später geglättet); thrombozytopenische Purpura; im roten Knochenmark befinden sich defekte Megakaryozyten; Manchmal werden Anomalien der Nierenentwicklung und angeborene Herzfehler festgestellt.
Symptome (Anzeichen)
Das klinische Bild wird durch die Grunderkrankung bestimmt, die die Thrombozytopenie verursacht hat.
Diagnose
Diagnostik Thrombozytopenie ist eine Indikation für die Untersuchung des Knochenmarks auf das Vorhandensein von Megakaryozyten. Ihr Fehlen weist auf eine Verletzung der Thrombozytopoese hin, und ihr Vorhandensein weist entweder auf eine Zerstörung der peripheren Blutplättchen oder (bei Vorliegen einer Splenomegalie) auf eine Ablagerung von Blutplättchen in der Milz hin. Pathologie der Thrombozytopoese. Die Diagnose wird durch den Nachweis einer megakaryozytären Dysplasie im Knochenmarksabstrich gesichert. Anomalien in der Bildung des Thrombozytenpools. Die Diagnose Hypersplenismus wird gestellt, wenn im Knochenmarkausstrich eine mäßige Thrombozytopenie mit einer normalen Anzahl von Megakaryozyten und einer deutlichen Vergrößerung der Milz festgestellt wird. Die Diagnose einer idiopathischen thrombozytopenischen Purpura erfordert den Ausschluss von Erkrankungen, die mit einer Thrombozytopenie einhergehen (z. B. SLE). und Thrombozytopenie, die durch die Einnahme von Medikamenten (z. B. Chinidin) verursacht wird. Verfügbare, aber unspezifische Methoden zum Nachweis von Thrombozytenaggregationshemmern sind bekannt.
Behandlung
Pathologie der Thrombozytopoese. Die Behandlung basiert, wenn möglich, auf der Beseitigung des auslösenden Erregers oder auf der Behandlung der Grunderkrankung; Die Halbwertszeit der Blutplättchen ist normalerweise normal, so dass Blutplättchentransfusionen bei Vorliegen einer Thrombozytopenie und Blutungszeichen möglich sind. Eine durch einen Mangel an Vitamin B 12 oder Folsäure verursachte Thrombozytopenie verschwindet mit der Wiederherstellung ihrer normalen Werte.
Amegakaryozytäre Thrombozytopenie spricht gut auf die Behandlung an; in der Regel werden Antithymozyten-Immunglobulin und Ciclosporin verschrieben.
Anomalien in der Bildung des Thrombozytenpools. Normalerweise gibt es keine Behandlung, obwohl eine Splenektomie das Problem lösen kann. Bei Transfusionen lagern sich einige Blutplättchen ab, wodurch Transfusionen weniger wirksam sind als bei verminderter Knochenmarksaktivität.
Behandlung der idiopathischen thrombozytopenischen Purpura – siehe Idiopathische thrombozytopenische Purpura.
Komplikationen und damit verbundene Erkrankungen Eine verminderte Thrombozytenproduktion geht mit aplastischer Anämie, Myelophthise (Ersatz von Knochenmark durch Tumorzellen oder fibröses Gewebe) und einigen seltenen angeborenen Syndromen ein Evans-Syndrom (Fisher-Evans-Syndrom) – eine Kombination aus autoimmunhämolytischer Anämie und autoimmuner Thrombozytopenie.
ICD-10 D69 Purpura und andere hämorrhagische Erkrankungen
Kodierung der Thrombozytopenie nach ICD 10
Blutplättchen spielen eine wichtige Rolle im menschlichen Körper und sind eine Gruppe von Blutzellen.
- 0 – Purpura, verursacht durch eine allergische Reaktion;
- 1 – Defekte in der Struktur von Blutplättchen mit normaler Anzahl;
- 2 – Purpura anderen, nicht thrombozytopenischen Ursprungs (im Falle einer Vergiftung);
- 3 – idiopathische thrombozytopenische Purpura;
- 4 – andere primäre Thrombozytendefizite;
- 5 – sekundäre Läsionen;
- 6 – nicht näher bezeichnete Varianten von Pathologien;
- 7 – andere Arten von Blutungen (Pseudogemophilie, erhöhte Brüchigkeit der Blutgefäße usw.);
- 8 – nicht näher bezeichnete hämorrhagische Zustände.
Diese Krankheitsgruppe wird unter der Überschrift Pathologien des Blutes, der hämatopoetischen Organe und Immunstörungen zellulären Ursprungs zusammengefasst.
Gefahr einer Thrombozytopenie
Aufgrund der Schwere der klinischen Manifestationen enthält Thrombozytopenie in der internationalen Klassifikation von Krankheiten Notfallprotokolle für schwere hämorrhagische Syndrome.
Auch beim Auftreten von Kratzern besteht Lebensgefahr mit einem starken Rückgang der Blutplättchenzahl, da die Wunde nicht durch primäre Blutgerinnsel heilt und weiter blutet.
Menschen mit einem Mangel an weißen Blutkörperchen können an spontanen inneren Blutungen sterben, daher erfordert die Krankheit eine rechtzeitige Diagnose und eine angemessene Behandlung.
Kommentar hinzufügen Antwort abbrechen
- Gepostet auf Akute Gastroenteritis
Selbstmedikation kann gesundheitsgefährdend sein. Konsultieren Sie bei den ersten Anzeichen einer Erkrankung einen Arzt.
Sekundäre Thrombozytopenie
Die Medikamente, die am häufigsten eine Thrombozytopenie verursachen, sind in der Tabelle aufgeführt. 16.5.
Heparin-induzierte Thrombozytopenie ist eine Heparin-induzierte, immunvermittelte prothrombotische Erkrankung, die mit Thrombozytopenie und venöser und/oder arterieller Thrombose einhergeht.
Ungefähr 1 % der Patienten entwickeln mindestens eine Woche nach der Heparinanwendung eine Heparin-induzierte Thrombozytopenie, und etwa 50 % davon erleiden eine Thrombose. Eine Heparin-induzierte Thrombozytopenie kommt bei Frauen etwas häufiger vor.
Ätiologie und Pathogenese
Eine Heparin-induzierte Thrombozytopenie resultiert aus einer humoralen Immunreaktion, die gegen einen Komplex aus endogenem Thrombozytenfaktor 4 und exogenem Heparin gerichtet ist; Autoantikörper erkennen endogenen Thrombozytenfaktor 4 nur, wenn er mit Heparin kombiniert wird. Dieser Immunkomplex aktiviert zirkulierende Blutplättchen über ihre FcγRIIA-Rezeptoren an der Oberfläche, was zu Thrombozytopenie und Hyperkoagulabilität führt. Die Eigenschaften von Heparin (Rind > Schwein), seine Zusammensetzung (unfraktioniert > niedriges Molekulargewicht > Fondaparinux), Dosis (prophylaktisch > therapeutisch > einmalig), Verabreichungsweg (subkutan > intravenös) und Dauer der Verabreichung (mehr als 4 Tage > weniger). als 4 Tage) sind alles Faktoren, die die Entwicklung und den Schweregrad einer Thrombozytopenie bestimmen.
Klinische Manifestationen
Bei medikamenteninduzierter Thrombozytopenie treten Petechien, Magen-Darm-Blutungen und Hämaturie meist mehrere Stunden nach der Medikamentengabe auf. Die Dauer einer Thrombozytopenie hängt von der Geschwindigkeit der Arzneimittelelimination ab. Normalerweise normalisiert sich die Thrombozytenzahl 7 Tage nach dem Absetzen wieder.
Eine Heparin-induzierte Thrombozytopenie kann in jedem Alter (> 3 Monate) auftreten, bei Kindern sind Fälle jedoch selten. Eine mittelschwere Thrombozytopenie beginnt normalerweise 5–10 Tage nach der Heparinverabreichung. Wenn der Patient in den letzten 100 Tagen bereits Heparin ausgesetzt war, kann es zu einer schnellen Reaktion kommen, wobei es innerhalb von Minuten bis Stunden nach der Heparinverabreichung zu einem Abfall der Thrombozytenzahl kommt. Auch eine verzögerte Heparin-induzierte Thrombozytopenie ist möglich; nach Absetzen des Arzneimittels entwickelt sich eine Thrombozytopenie. Thrombozytopenie verläuft normalerweise asymptomatisch und Blutungen sind selten. Eine Heparin-induzierte Thrombozytopenie ist mit einem hohen Risiko für thrombotische Komplikationen (z. B. Lungenembolie, Myokardinfarkt, thrombotischer Schlaganfall) verbunden, mit einer starken Vorliebe für arterielle Thrombosen der Extremitätenarterien und tiefe Venenthrombosen. Zusätzliche mikrovaskuläre Thrombosen können zur Entwicklung einer venösen Gangrän/Amputation von Gliedmaßen führen. Weitere Komplikationen sind Hautnekrosen an Heparin-Injektionsstellen und anaphylaktoide Reaktionen (z. B. Fieber, Hypotonie, Gelenkschmerzen, Atemnot, Herz-Lungen-Versagen) nach intravenöser Bolusverabreichung.
Sekundäre Thrombozytopenie: Diagnose
Die Diagnose einer Heparin-induzierten Thrombozytopenie kann aufgrund des klinischen Bildes vermutet werden – Thrombozytopenie, Thrombose, Fehlen einer anderen Ursache der Thrombozytopenie. Die Diagnose wird durch den Nachweis von Antikörpern gegen den endogenen Thrombozytenfaktor 4/Heparin-Komplex und durch den Nachweis pathologischer Thrombozyten-aktivierender Antikörper mittels eines Serotonin-Freisetzungstests oder eines Heparin-induzierten Thrombozytenaktivierungstests bestätigt.
Differentialdiagnose
Zu den Differenzialdiagnosen gehören eine nicht-immune Heparin-assoziierte Thrombozytopenie (aufgrund der direkten Wechselwirkung von Heparin mit zirkulierenden Blutplättchen in den ersten Tagen nach der Heparin-Gabe) sowie postoperative Hämodilution, Sepsis, nicht-Heparin-induzierte Thrombozytopenie, disseminierte intravaskuläre Koagulation, und Multiorganversagen.
Sekundäre Thrombozytopenie: Behandlung
Bei einigen Patienten, die Heparin erhalten, wird eine regelmäßige Überwachung der Thrombozytenzahl empfohlen. Wenn eine Heparin-induzierte Thrombozytopenie vermutet oder bestätigt wird, besteht die Behandlung darin, Heparin abzusetzen und ein alternatives Antikoagulans zu verwenden, entweder Nicht-Heparin-Anti-Faktor Xa (Danaparoid, Fondaparinux) oder direkte Thrombininhibitoren (z. B. Argatroban, Bivalirudin). Warfarin ist während der akuten thrombozytopenischen Phase kontraindiziert, da es eine mikrovaskuläre Thrombose mit der Möglichkeit einer Nekrose der ischämischen Extremität (venöses Gangrän-Syndrom) verursachen kann. Eine Thrombozytopenie verschwindet in der Regel nach durchschnittlich 4 Tagen bei Werten über 150 x 109/L, in manchen Fällen kann es jedoch auch 1 Woche bis 1 Monat dauern.
Die Prognose für die Erholung der Thrombozytenzahl ist gut, es können jedoch postthrombotische Komplikationen auftreten (z. B. Gliedmaßenamputation bei 5–10 % der Patienten, Schlaganfall, beidseitige hämorrhagische Nebennierennekrose mit Nebenniereninsuffizienz). In 5–10 % der Fälle kommt es zu einer Mortalität aufgrund einer Heparin-induzierten Thrombozytopenie (z. B. tödliche Lungenembolie).
Prävention
Andere [Bearbeiten]
Thrombozytopenische Purpura, verursacht durch Transfusion roter Blutkörperchen
1. Klinisches Bild. Thrombozytopenische Purpura ist eine seltene Komplikation einer Transfusion roter Blutkörperchen. Sie äußert sich in einer plötzlichen Thrombozytopenie, Blutungen aus den Schleimhäuten und Petechien, die 7–10 Tage nach der Transfusion auftreten. Die Diagnose basiert auf der Krankengeschichte. Diese Form der thrombozytopenischen Purpura tritt am häufigsten bei Mehrgebärenden und bei Menschen auf, die sich mehreren Transfusionen roter Blutkörperchen unterzogen haben. Vom Entwicklungsmechanismus her ähnelt es der Thrombozytopenie bei Neugeborenen, die durch mütterliche Antikörper verursacht wird. Thrombozytopenische Purpura, die durch eine Transfusion roter Blutkörperchen verursacht wird, tritt bei Personen auf, denen das Zw a-Antigen fehlt. Es wurde gezeigt, dass dieses Antigen Teil des Glykoproteins IIb/IIIa ist. Die Transfusion roter Blutkörperchen, gemischt mit Blutplättchen, die das Zw a-Antigen tragen, führt zum Auftreten von Antikörpern gegen dieses Antigen. Es wird angenommen, dass sie mit dem Glykoprotein IIb/IIIa der eigenen Blutplättchen des Patienten kreuzreagieren.
A. Blutplättchentransfusionen werden nicht durchgeführt, da sie in der Regel wirkungslos sind. Darüber hinaus können bei dieser Krankheit nur 2 % der Menschen Blutplättchen spenden, deren Blutplättchen nicht das Zw a-Antigen tragen.
B. Prednison, 1–2 mg/kg/Tag oral, reduziert das hämorrhagische Syndrom und erhöht die Thrombozytenzahl.
V. Die Krankheit verschwindet von selbst, nachdem das Blut des Patienten von den Blutplättchen des Spenders befreit wurde.
d. Anschließend sollten rote Blutkörperchen von Spendern, denen das Zw a-Antigen fehlt, zur Transfusion verwendet werden.
Thrombozytopenie: Symptome und Behandlung
Thrombozytopenie – Hauptsymptome:
- Rote Flecken auf der Haut
- Vergrößerte Lymphknoten
- Fieber
- Vergrößerte Lymphknoten im Nacken
- Leichte Blutungen an Haut und Schleimhäuten
- Blaue Flecken auf der Haut
Eine Krankheit, die zu einer Verringerung der Anzahl der Blutplättchen im Blut führt, wird Thrombozytopenie genannt. Genau darüber wird in dem Artikel gesprochen. Blutplättchen sind kleine Blutzellen, die keine Farbe haben und wichtige Bestandteile der Blutgerinnung sind. Die Krankheit ist ziemlich schwerwiegend, da die Krankheit zu Blutungen in den inneren Organen (insbesondere im Gehirn) führen kann, die tödlich sind.
Einstufung
Wie die meisten medizinischen Erkrankungen gibt es auch für die Thrombozytopenie eine eigene Klassifikation, die auf der Grundlage pathogenetischer Faktoren, Ursachen, Symptome und verschiedener Erscheinungsformen gebildet wird.
Nach dem Kriterium der Ätiologie werden zwei Krankheitstypen unterschieden:
Sie zeichnen sich dadurch aus, dass sich der primäre Typ als eigenständige Krankheit manifestiert und der sekundäre Typ durch eine Reihe anderer Krankheiten oder pathologischer Anomalien hervorgerufen wird.
Je nach Dauer der Krankheit im menschlichen Körper gibt es zwei Arten von Unwohlsein: akutes und chronisches. Akut – gekennzeichnet durch eine kurze Einwirkungsdauer auf den Körper (bis zu sechs Monate), die sich jedoch durch sofortige Symptome äußert. Die chronische Form ist durch eine anhaltende Abnahme der Blutplättchen im Blut (über sechs Monate) gekennzeichnet. Gefährlicher ist der chronische Typ, da die Behandlung bis zu zwei Jahre dauert.
Nach den Kriterien für die Schwere der Erkrankung, die durch die quantitative Zusammensetzung der Blutplättchen im Blut gekennzeichnet sind, werden drei Grade unterschieden:
- I – die Zusammensetzung beträgt 150–50 x 10 9 /l – das Schwerekriterium ist zufriedenstellend;
- II – 50–20 x 10 9 /l – reduzierte Zusammensetzung, die sich in leichten Hautschäden äußert;
- III – 20 x 10 9 /l – gekennzeichnet durch das Auftreten innerer Blutungen im Körper.
Die Norm der Blutzellen im Körper beträgt 0,00/µl. Aber gerade im weiblichen Körper ändern sich diese Indikatoren ständig. Die Veränderungen werden durch folgende Faktoren beeinflusst:
Blutplättchen entstehen im Körper aus dem Knochenmark, das durch die Stimulation von Megakaryozyten Blutzellen synthetisiert. Die synthetisierten Blutplättchen zirkulieren sieben Tage lang durch das Blut, danach wiederholt sich der Prozess ihrer Stimulation.
Gemäß der Internationalen Klassifikation der Krankheiten der Zehnten Versammlung (ICD-10) hat diese Krankheit ihre eigenen Codes:
- D50-D89 – Erkrankungen des Kreislaufsystems und andere Arten von Insuffizienz.
- D65-D69 – Blutgerinnungsstörungen.
Ursachen
Ursache der Erkrankung ist häufig eine allergische Reaktion des Körpers auf verschiedene Medikamente, die zu einer medikamenteninduzierten Thrombozytopenie führt. Bei einer solchen Erkrankung produziert der Körper Antikörper gegen das Medikament. Zu den Arzneimitteln, die das Auftreten einer Blutzellinsuffizienz beeinflussen, gehören Beruhigungsmittel, Alkaloide und antibakterielle Wirkstoffe.
Ursachen für einen Mangel können auch Probleme des Immunsystems sein, die durch die Folgen von Bluttransfusionen verursacht werden.
Besonders häufig äußert sich die Erkrankung dann, wenn die Blutgruppen nicht übereinstimmen. Autoimmunthrombozytopenie wird am häufigsten im menschlichen Körper beobachtet. In diesem Fall ist das Immunsystem nicht in der Lage, die Blutplättchen zu erkennen und stößt sie aus dem Körper aus. Als Folge der Abstoßung werden Antikörper produziert, um fremde Zellen zu entfernen. Die Ursachen einer solchen Thrombozytopenie sind:
- Pathologisches Nierenversagen und chronische Hepatitis.
- Lupus, Dermatomyositis und Sklerodermie.
- Leukämische Erkrankungen.
Wenn es sich bei der Erkrankung um eine ausgeprägte Form einer isolierten Erkrankung handelt, spricht man von idiopathischer Thrombozytopenie oder Werlhof-Krankheit (ICD-10-Code: D69.3). Die Ätiologie der idiopathischen thrombozytopenischen Purpura (ICD-10:D63.6) bleibt unklar, Mediziner neigen jedoch zu der Annahme, dass die Ursache eine erbliche Veranlagung ist.
Typisch ist die Manifestation der Erkrankung auch bei Vorliegen einer angeborenen Immunschwäche. Solche Menschen sind am anfälligsten für die Faktoren, die die Krankheit verursachen. Die Gründe dafür sind:
- Schädigung des roten Knochenmarks durch Drogeneinfluss;
- Eine Immunschwäche führt zu einer Schädigung der Megakaryozyten.
Die Krankheit hat einen produktiven Charakter, der durch eine unzureichende Produktion von Blutplättchen im Knochenmark verursacht wird. In diesem Fall kommt es zu einer Unzulänglichkeit, die sich letztendlich zu einem Unwohlsein entwickelt. Als Ursachen gelten Myelosklerose, Metastasen, Anämie etc.
Bei Menschen mit einer verminderten Zusammensetzung von Vitamin B12 und Folsäure wird ein Mangel an Blutplättchen im Körper beobachtet. Es kann nicht ausgeschlossen werden, dass eine übermäßige radioaktive oder Strahlenexposition zu einem Mangel an Blutzellen führt.
Somit können zwei Arten von Ursachen unterschieden werden, die das Auftreten einer Thrombozytopenie beeinflussen:
- Führt zur Zerstörung von Blutzellen: idiopathische thrombozytopenische Purpura, Autoimmunerkrankungen, Herzoperationen, klinische Durchblutungsstörungen bei Schwangeren und Nebenwirkungen von Medikamenten.
- Zu einer Verringerung der Antikörperproduktion des Knochenmarks beitragen: virale Einflüsse, metastatische Manifestationen, Chemotherapie und Bestrahlung sowie übermäßiger Alkoholkonsum.
Symptome
Die Symptome einer Thrombozytopenie-Erkrankung können unterschiedliche Erscheinungsformen haben. Es kommt darauf an:
- erstens aus der Ursache des Ereignisses;
- zweitens von der Art der Erkrankung (chronisch oder akut).
Die wichtigsten Anzeichen einer Schädigung des Körpers sind Erscheinungen auf der Haut in Form von Blutungen und Blutungen. Blutungen werden am häufigsten an den Gliedmaßen und am Rumpf beobachtet. Schäden am Gesicht und an den Lippen einer Person sind möglich. Um die Manifestation von Blutungen am menschlichen Körper zu veranschaulichen, wird das folgende Foto dargestellt.
Thrombozytopenie ist durch Symptome längerer Blutungen nach Zahnextraktion gekennzeichnet. Darüber hinaus kann die Dauer der Blutung entweder einen Tag betragen oder sich über mehrere Tage erstrecken. Es kommt auf das Ausmaß der Erkrankung an.
Bei Symptomen kommt es nicht zu einer Vergrößerung der Leber, sehr oft beobachten Ärzte jedoch eine Vergrößerung der Lymphknoten der Halswirbelsäule. Dieses Phänomen geht häufig mit einem Anstieg der Körpertemperatur auf subfebrile Werte (von 37,1 auf 38 Grad) einher. Ein Anstieg der Ansammlung roter Blutkörperchen im Körper ist ein Hinweis auf das Vorliegen einer Krankheit namens Lupus erythematodes.
Symptome eines Thrombozytenmangels sind nach der Blutentnahme zur Analyse recht leicht zu beobachten. Die quantitative Zusammensetzung wird deutlich von den Höchststandards abweichen. Wenn die Anzahl der Blutplättchen im Blut abnimmt, nimmt ihre Größe zu. Dies spiegelt sich auf der Haut durch das Auftreten roter und bläulicher Flecken wider, die auf die Umwandlung von Blutzellen hinweisen. Es kommt auch zu einer Zerstörung der roten Blutkörperchen, was zu einer Verringerung der quantitativen Zusammensetzung führt, gleichzeitig aber die Anzahl der Retikulozyten zunimmt. Es besteht das Phänomen einer Verschiebung der Leukozytenformel nach links.
Der menschliche Körper mit einer reduzierten Zusammensetzung der Blutzellen ist durch eine Zunahme der Zusammensetzung der Megakaryozyten gekennzeichnet, die durch häufige und starke Blutungen verursacht wird. Die Dauer der Blutgerinnung wird spürbar verlängert und die Gerinnungsminderung des aus der Wunde freigesetzten Blutes wird verringert.
Je nach Krankheitssymptom werden drei Schweregrade der Komplikationen unterschieden: leicht, mittelschwer und schwer.
Leichte Verläufe sind typische Krankheitsursachen bei Frauen mit längerer und starker Menstruation sowie mit intradermalen Blutungen und Nasenbluten. Im milden Stadium ist die Diagnose jedoch äußerst schwierig, sodass das Vorliegen der Krankheit erst nach einer ausführlichen ärztlichen Untersuchung bestätigt werden kann.
Der durchschnittliche Grad ist durch das Auftreten eines hämorrhagischen Ausschlags am ganzen Körper gekennzeichnet, der aus zahlreichen punktförmigen Blutungen unter der Haut und auf der Schleimhaut besteht.
Schwere Grade sind durch gastrointestinale Störungen gekennzeichnet, die durch Blutungen verursacht werden. Die Thrombozytenzahl im Blut beträgt bis zu 25x10 9 /l.
Die Symptome einer sekundären Thrombozytopenie weisen ähnliche Merkmale auf.
Schwangerschaft und Krankheit: Symptome
Thrombozytopenie bei schwangeren Frauen ist durch signifikante Veränderungen in der quantitativen Zusammensetzung der Zellen im Blut von Frauen gekennzeichnet. Wenn bei schwangeren Frauen keine Krankheitsdiagnose vorliegt, der Indikator für die Blutplättchenzusammensetzung jedoch leicht abnimmt, deutet dies darauf hin, dass ihre Vitalaktivität abnimmt und ihre Beteiligung an der Peripherie des Blutkreislaufs zunimmt.
Liegt im Blut einer Schwangeren eine verminderte Blutplättchenzusammensetzung vor, sind dies unmittelbare Voraussetzungen für die Entstehung der Erkrankung. Die Gründe für die verringerte Anzahl von Blutplättchen sind die hohe Absterberate dieser Körper und die geringe Neubildungsrate. Klinische Symptome sind durch subkutane Blutungen gekennzeichnet. Die Ursachen für einen Mangel an farblosen Zellen sind falsche Zusammensetzung und Ernährungsstandards oder eine geringe Nahrungsaufnahme sowie eine Schädigung des Immunsystems und verschiedene Blutverluste. Durch diesen Prozess werden die Blutkörperchen vom Knochenmark in geringen Mengen produziert oder weisen eine unregelmäßige Form auf.
Thrombozytopenie während der Schwangerschaft ist sehr gefährlich, daher wird der Frage der Diagnose und insbesondere der Behandlung größte Aufmerksamkeit geschenkt. Die Gefahr besteht darin, dass ein Mangel an Blutplättchen im Blut der Mutter während der Schwangerschaft zu Blutungen beim Kind führt. Die gefährlichste Blutung im Mutterleib ist die Gehirnblutung, deren Ergebnis tödliche Folgen für den Fötus hat. Beim ersten Anzeichen eines solchen Faktors entscheidet der Arzt über eine Frühgeburt, um die Folgen zu beseitigen.
Thrombozytopenie im Kindesalter: Symptome
Thrombozytopenie bei Kindern ist recht selten. Zur Risikogruppe zählen Kinder im schulpflichtigen Alter, deren Inzidenz häufiger im Winter und Frühjahr auftritt.
Thrombozytopenie und ihre Symptome unterscheiden sich bei Kindern praktisch nicht von denen bei Erwachsenen, es ist jedoch wichtig, dass Eltern sie anhand der ersten Anzeichen in den frühen Stadien der Krankheitsentwicklung diagnostizieren. Zu den Symptomen bei Kindern zählen häufige Blutungen aus der Nasenhöhle und das Auftreten kleiner Ausschläge am Körper. Der Ausschlag tritt zunächst an den unteren Extremitäten des Körpers auf und kann dann an den Armen beobachtet werden. Bei leichten Prellungen kommt es zu Schwellungen und Hämatomen. Solche Anzeichen bereiten den Eltern meist keinen Anlass zur Sorge, da keine Schmerzsymptome vorliegen. Dies ist ein wichtiger Fehler, da jede Krankheit in ihrer fortgeschrittenen Form gefährlich ist.
Zahnfleischbluten weist sowohl bei Kindern als auch bei Erwachsenen auf einen Mangel an Blutplättchen im Blut hin. In diesem Fall wird der Kot bei einer kranken Person und häufiger bei Kindern zusammen mit Blutgerinnseln ausgeschieden. Eine Blutung durch Wasserlassen kann nicht ausgeschlossen werden.
Je nach Ausmaß der Beeinträchtigung des Immunsystems durch die Erkrankung wird zwischen immunologischem und nichtimmunologischem Thrombozytenmangel unterschieden. Eine Immunthrombozytopenie wird durch das massive Absterben von Blutzellen unter dem Einfluss von Antikörpern verursacht. In einer solchen Situation werden die körpereigenen Blutzellen des Immunsystems nicht erkannt und vom Körper abgestoßen. Nicht-immunologische Erkrankungen äußern sich durch körperliche Auswirkungen auf die Blutplättchen.
Diagnose
Eine Person sollte bei den ersten Anzeichen und Symptomen der Krankheit diagnostiziert werden. Die wichtigste Diagnosemethode ist ein klinischer Bluttest, dessen Ergebnisse ein Bild der quantitativen Zusammensetzung der Blutplättchen zeigen.
Wird eine Abweichung in der Anzahl der Blutzellen im Körper festgestellt, besteht die Indikation für eine Knochenmarksuntersuchung. Somit wird das Vorhandensein von Megakaryozyten festgestellt. Fehlen sie, ist die Thrombusbildung beeinträchtigt und ihr Vorhandensein weist auf die Zerstörung von Blutplättchen oder deren Ablagerung in der Milz hin.
Die Ursachen eines Mangels werden diagnostiziert durch:
- Gentests;
- Elektrokardiogramme;
- Tests auf das Vorhandensein von Antikörpern;
- Ultraschalluntersuchungen;
- Röntgen und Endoskopie.
Die Diagnose einer Thrombozytopenie während der Schwangerschaft erfolgt mithilfe eines Koagulogramms, vereinfacht ausgedrückt, eines Blutgerinnungstests. Mit dieser Analyse können Sie die Zusammensetzung der Blutplättchen im Blut genau bestimmen. Der Verlauf des Geburtsvorgangs hängt von der Anzahl der Blutplättchen ab.
Behandlung
Die Behandlung einer Thrombozytopenie beginnt mit einer Therapie, bei der im Krankenhaus ein Medikament namens Prednisolon verschrieben wird.
Wichtig! Behandlungsmethoden werden vom behandelnden Arzt nur nach entsprechender Untersuchung und Diagnose der Krankheit streng verordnet.
Die Dosierung des Medikaments ist in der Gebrauchsanweisung angegeben, wonach 1 ml des Medikaments pro 1 kg Körpergewicht eingenommen wird. Mit fortschreitender Krankheit erhöht sich die Dosis um das 1,5- bis 2-fache. Im Anfangsstadium zeichnet sich die Erkrankung durch eine schnelle und wirksame Genesung aus, so dass Sie nach Einnahme des Arzneimittels innerhalb weniger Tage eine Verbesserung des Gesundheitszustandes feststellen können. Die Einnahme des Arzneimittels wird so lange fortgesetzt, bis die Person vollständig geheilt ist, was vom behandelnden Arzt bestätigt werden muss.
Die Wirkung von Glukokortikosteroiden wirkt sich positiv auf die Bekämpfung von Unwohlsein aus, in den meisten Fällen verschwinden jedoch nur die Symptome und die Krankheit bleibt bestehen. Zur Behandlung von Mangelerscheinungen bei Kindern und Jugendlichen.
Die Behandlung der idiopathischen chronischen Thrombozytopenie erfolgt durch Entfernung der Milz. Dieser Eingriff wird in der Medizin als Splenektomie bezeichnet und zeichnet sich durch seine positiven Wirkungen aus. Vor der Operation wird die Dosierung von Prednisolon um das Dreifache erhöht. Darüber hinaus wird es nicht in einen Muskel, sondern direkt in eine menschliche Vene injiziert. Nach der Splenektomie wird die Verabreichung des Arzneimittels in den gleichen Dosen bis zu zwei Jahre lang fortgesetzt. Erst nach Ablauf der vorgegebenen Frist erfolgt eine Untersuchung und Bescheinigung über den Erfolg der Splenektomie.
Bleibt die Entfernungsoperation erfolglos, wird dem Patienten eine immunsuppressive Chemotherapie mit Zytostatika verschrieben. Zu diesen Medikamenten gehören: Azathioprin und Vincristin.
Wenn ein erworbener Mangel nichtimmuner Natur diagnostiziert wird, wird die Thrombozytopenie symptomatisch durch die Einnahme von Östrogenen, Gestagenen und Androxonen behandelt.
Schwerwiegendere Formen der idiopathischen Thrombozytopenie werden durch übermäßige Blutungen verursacht. Zur Wiederherstellung des Blutes wird eine Transfusion durchgeführt. Die Behandlung schwerer Fälle erfordert das Absetzen von Medikamenten, die sich negativ auf die Fähigkeit der Blutplättchen zur Gerinnselbildung auswirken können.
Nach der Diagnose der Krankheit wird der Patient registriert und es findet eine Untersuchung nicht nur des Patienten, sondern auch seiner Angehörigen statt, um eine erbliche Vorgeschichte zu erheben.
Bei Kindern lässt sich das Unwohlsein gut und komplikationslos behandeln, in manchen Fällen ist jedoch eine symptomatische Therapie nicht auszuschließen.
Auch die Behandlung der Thrombozytopenie mit der traditionellen Medizin hat beachtliche Erfolge erzielt. Um das Problem des Blutplättchenmangels zu beseitigen, sollten Sie zunächst Honig und Walnüsse in Ihre Ernährung aufnehmen. Auch Abkochungen aus Brennnessel- und Hagebuttenblättern helfen gut. Zur Vorbeugung wird Birken-, Himbeer- oder Rübensaft verwendet.
Wenn Sie glauben, an einer Thrombozytopenie und den für diese Krankheit charakteristischen Symptomen zu leiden, kann Ihnen ein Hämatologe helfen.
Wir empfehlen außerdem die Nutzung unseres Online-Krankheitsdiagnosedienstes, der anhand der eingegebenen Symptome wahrscheinliche Krankheiten auswählt.
Diphtherie ist eine Infektionskrankheit, die durch den Kontakt mit einem bestimmten Bakterium hervorgerufen wird und dessen Übertragung (Infektion) durch Tröpfchen in der Luft erfolgt. Diphtherie, deren Symptome die Aktivierung des Entzündungsprozesses hauptsächlich im Nasopharynx und Oropharynx sind, ist auch durch begleitende Manifestationen in Form einer allgemeinen Vergiftung und einer Reihe von Läsionen gekennzeichnet, die sich direkt auf das Ausscheidungs-, Nerven- und Herz-Kreislauf-System auswirken.
Masern sind eine akute Infektionskrankheit, deren Anfälligkeitsgrad nahezu 100 % beträgt. Masern, zu deren Symptomen Fieber, ein entzündlicher Prozess der Schleimhäute der Mundhöhle und der oberen Atemwege, das Auftreten eines makulopapulösen Ausschlags auf der Haut, eine allgemeine Vergiftung und eine Bindehautentzündung gehören, sind eine der Haupttodesursachen bei jungen Menschen Kinder.
Leptospirose ist eine Infektionskrankheit, die durch bestimmte Erreger der Gattung Leptospira verursacht wird. Der pathologische Prozess betrifft vor allem die Kapillaren sowie Leber, Nieren und Muskeln.
Pharyngomykose (Tonsillomykose) ist eine Pathologie der Rachenschleimhaut akuter oder chronischer Natur, deren Hauptursache eine Infektion des Körpers durch Pilze ist. Pharyngomykose betrifft Menschen aller Altersgruppen, darunter auch kleine Kinder. Selten tritt die Erkrankung isoliert auf.
Toxisches Erythem ist eine Krankheit, bei deren Fortschreiten ein polymorpher Ausschlag auf der menschlichen Haut auftritt. Die Krankheit betrifft am häufigsten Neugeborene, ein Auftreten bei erwachsenen Patienten ist jedoch möglich. Bei 50 % der Babys entwickelt sich in den ersten Lebenstagen ein Erythema toxicum des Neugeborenen. Dieser Zustand spiegelt den Anpassungsprozess des Kindes an die Umwelt sowie an äußere Faktoren wider.
Mit Hilfe von Bewegung und Abstinenz können die meisten Menschen auf Medikamente verzichten.
Symptome und Behandlung menschlicher Krankheiten
Die Vervielfältigung von Materialien ist nur mit Genehmigung der Verwaltung und Angabe eines aktiven Links zur Quelle möglich.
Alle Angaben unterliegen der zwingenden Rücksprache mit Ihrem behandelnden Arzt!
Fragen und Anregungen:
ICD-Code: D69.6
Thrombozytopenie, nicht näher bezeichnet
Thrombozytopenie, nicht näher bezeichnet
Suchen
- Suche nach ClassInform
Durchsuchen Sie alle Klassifikatoren und Nachschlagewerke auf der ClassInform-Website
Suche nach TIN
- OKPO von TIN
Suchen Sie den OKPO-Code nach INN
Suchen Sie den OKTMO-Code nach INN
Suchen Sie den OKATO-Code nach INN
Suchen Sie den OKOPF-Code anhand der TIN
Suchen Sie den OKOGU-Code nach INN
Suchen Sie nach OKFS-Code anhand der TIN
Suchen Sie nach OGRN anhand der TIN
Suchen Sie nach der TIN einer Organisation nach Namen, der TIN eines einzelnen Unternehmers nach vollständigem Namen
Überprüfung der Gegenpartei
- Überprüfung der Gegenpartei
Informationen zu Gegenparteien aus der Datenbank des Federal Tax Service
Konverter
- OKOF bis OKOF2
Übersetzung des OKOF-Klassifikatorcodes in den OKOF2-Code
Übersetzung des OKDP-Klassifikatorcodes in OKPD2-Code
Übersetzung des OKP-Klassifikatorcodes in OKPD2-Code
Übersetzung des OKPD-Klassifikatorcodes (OK(KPES 2002)) in den OKPD2-Code (OK(KPES 2008))
Übersetzung des OKUN-Klassifikatorcodes in OKPD2-Code
Übersetzung des OKVED2007-Klassifikatorcodes in den OKVED2-Code
Übersetzung des OKVED2001-Klassifikatorcodes in den OKVED2-Code
Übersetzung des OKATO-Klassifikatorcodes in OKTMO-Code
Übersetzung des HS-Codes in den OKPD2-Klassifikatorcode
Übersetzung des OKPD2-Klassifikatorcodes in den HS-Code
Übersetzung des OKZ-93-Klassifikatorcodes in den OKZ-2014-Code
Klassifikatoränderungen
- Änderungen 2018
Feed der in Kraft getretenen Klassifikatoränderungen
Allrussische Klassifikatoren
- ESKD-Klassifikator
Allrussischer Klassifikator für Produkte und Designdokumente OK
Allrussischer Klassifikator von Objekten der administrativ-territorialen Aufteilung OK
Allrussischer Währungsklassifikator OK (MK (ISO 4))
Allrussischer Klassifikator für Frachtarten, Verpackungen und Verpackungsmaterialien OK
Allrussischer Klassifikator der Arten wirtschaftlicher Aktivitäten OK (NACE Rev. 1.1)
Allrussischer Klassifikator der Arten wirtschaftlicher Aktivitäten OK (NACE REV. 2)
Allrussischer Klassifikator der Wasserkraftressourcen OK
Allrussischer Klassifikator der Maßeinheiten OK(MK)
Allrussischer Berufsklassifikator OK (MSKZ-08)
Allrussischer Klassifikator von Informationen über die Bevölkerung OK
Allrussischer Klassifikator von Informationen zum sozialen Schutz der Bevölkerung. OK (gültig bis 01.12.2017)
Allrussischer Klassifikator von Informationen zum sozialen Schutz der Bevölkerung. OK (gültig ab 01.12.2017)
Allrussischer Klassifikator der beruflichen Grundbildung OK (gültig bis 01.07.2017)
Allrussischer Klassifikator der Regierungsbehörden OK 006 – 2011
Allrussischer Klassifikator mit Informationen über allrussische Klassifikatoren. OK
Allrussischer Klassifikator der Organisations- und Rechtsformen OK
Allrussischer Klassifikator des Anlagevermögens OK (gültig bis 01.01.2017)
Allrussischer Klassifikator des Anlagevermögens OK (SNA 2008) (gültig ab 01.01.2017)
Allrussischer Produktklassifikator OK (gültig bis 01.01.2017)
Allrussischer Produktklassifizierer nach Art der Wirtschaftstätigkeit OK (CPES 2008)
Allrussischer Klassifikator für Arbeitnehmerberufe, Arbeitnehmerpositionen und Tarifkategorien OK
Allrussischer Klassifikator für Mineralien und Grundwasser. OK
Allrussischer Klassifikator von Unternehmen und Organisationen. OK 007–93
Allrussischer Klassifikator der OK-Standards (MK (ISO/infko MKS))
Allrussischer Klassifikator für Fachgebiete mit höherer wissenschaftlicher Qualifikation OK
Allrussischer Klassifikator der Länder der Welt OK (MK (ISO 3))
Allrussischer Klassifikator der Fachrichtungen im Bildungsbereich OK (gültig bis 01.07.2017)
Allrussischer Klassifikator der Fachrichtungen im Bildungsbereich OK (gültig ab 01.07.2017)
Allrussischer Klassifikator für Transformationsereignisse OK
Allrussischer Klassifikator der Gemeindegebiete OK
Allrussischer Klassifizierer der Managementdokumentation OK
Allrussischer Klassifikator der Eigentumsformen OK
Allrussischer Klassifikator der Wirtschaftsregionen. OK
Allrussischer Klassifikator der Dienstleistungen für die Bevölkerung. OK
Warennomenklatur der Außenwirtschaftstätigkeit (EAEU CN FEA)
Klassifikator der Arten der zulässigen Nutzung von Grundstücken
Klassifizierung der Operationen des Sektors Staat
Bundesklassifizierungskatalog Abfall (gültig bis 24.06.2017)
Abfallklassifizierungskatalog des Bundes (gültig ab 24.06.2017)
Internationale Klassifikatoren
Universeller Dezimalklassifikator
Internationale Klassifikation der Krankheiten
Anatomisch-therapeutisch-chemische Klassifizierung von Arzneimitteln (ATC)
Internationale Klassifikation von Waren und Dienstleistungen, 11. Auflage
Internationale Klassifikation für Industriedesign (10. Revision) (LOC)
Verzeichnisse
Einheitliches Tarif- und Qualifikationsverzeichnis der Arbeiten und Berufe der Arbeitnehmer
Einheitliches Qualifikationsverzeichnis der Positionen von Führungskräften, Fachkräften und Mitarbeitern
Verzeichnis der Berufsstandards für 2017
Muster von Stellenbeschreibungen unter Berücksichtigung beruflicher Standards
Bildungsstandards der Bundesländer
Allrussische Stellendatenbank „Arbeiten in Russland“.
Staatliches Kataster der Zivil- und Dienstwaffen und Munition für sie
Produktionskalender für 2017
Produktionskalender für 2018
Idiopathische thrombozytopenische Purpura (ITP)- eine Krankheit unbekannter Ätiologie, die durch die Entwicklung einer Thrombozytopenie und eines hämorrhagischen Syndroms gekennzeichnet ist. Am häufigsten wird die Zerstörung der Blutplättchen durch einen Autoimmunprozess verursacht, der durch einen Infektionserreger oder die Einnahme von Medikamenten ausgelöst wird. Überwiegendes Alter- bis 14 Jahre alt. Vorherrschendes Geschlecht- weiblich.
Code gemäß der internationalen Klassifikation von Krankheiten ICD-10:
- D69.3
Ursachen
Genetische Aspekte. Es wurde eine hereditäre thrombozytopenische autoimmune idiopathische Purpura beschrieben (188030, B), die sich durch ein hämorrhagisches Syndrom, Thrombozytopenie und das Vorhandensein von Antikörpern gegen Blutplättchen äußert.
Einstufung. Je nach Verlauf: akut (weniger als 6 Monate), chronisch (mehr als 6 Monate). Phasen der Erkrankung. Phase der Exazerbation (Krise). Klinische Remission. Klinische und hämatologische Remission. Je nach Krankheitsbild... Trocken (vereinzelte Hauterscheinungen) .. Nass (Zusatz von Blutungen aus den Schleimhäuten).
Krankheitsbild
. Akuter Beginn mit hämorrhagischem Syndrom. Die Körpertemperatur kann auf subfebrile Werte ansteigen.
. Der Zustand des Patienten ändert sich oft nicht wesentlich.
. Petechial – ekchymotischer Ausschlag, lokalisiert am Gesäß, an den Innenseiten der Oberschenkel, auf der Brust und im Gesicht.
. Auch im Stadium der klinischen Remission ist ein positives Pinch-Symptom möglich.
. Blutungen aus Schleimhäuten. Am häufigsten kommt es zu starkem Nasenbluten; bei Mädchen in der Pubertät - Uterusblutungen.
. Innere Blutungen im Magen-Darm-Trakt und im Zentralnervensystem sind äußerst selten.
Laborforschung. Blutbild: posthämorrhagische Anämie, Thrombozytopenie. OAM – mögliche Hämaturie aufgrund von Nierenblutungen. Immunogramm: Anstieg des CEC-Gehalts. Myelogramm: „Reizung“ der Megakaryozytenlinie, Auftreten „inaktiver“ Megakaryozyten.
Behandlung
BEHANDLUNG
Modus Bettruhe bei schwerer Thrombozytopenie.
Diät mit Ausnahme der obligatorischen Allergene.
Drogen Therapie
. Bei Blutungen – Etamsylat, Aminocapronsäure, Carbazochrom, blutstillende Mittel zur topischen Anwendung, bei starkem Nasenbluten – Nasentamponade. Bei Uterusblutungen - Oxytocin (wie vom Gynäkologen verschrieben).
. Antihistaminika.
. Vitamin B15, Eleutherococcus.
. GC, zum Beispiel Prednisolon. Indikationen – feuchte Form der ITP, reichliche Hauterscheinungen im Gesicht, Kopfhaut mit einem Thrombozytengehalt von weniger als 0,051012/l. Verschrieben in Kursen von 2-3 mg/kg/Tag für 5-7 Tage mit Pausen von 5-7 Tagen. Indikation für einen Abbruch ist eine klinische und hämatologische Remission bis zum ersten Tag des nächsten Kurses. Wenn die Thrombozytopenie bestehen bleibt und kein hämorrhagisches Syndrom vorliegt, wird die Therapie nach 4-5 Kursen abgebrochen.
. Inosin; Orotsäure, Kaliumsalz; Liponsäure.
. Immunsuppressive Therapie – Wirksamkeit fraglich.
. Alternative Medikamente. Rekombinante IFN-Präparate für chronische Erkrankungen. Induktion: 3 Millionen Einheiten/m2 3 U/Woche. Die Dauer des Kurses richtet sich nach der „Reaktion“ (Erholungsphase und Thrombozytenzahl). Erhaltungstherapie für 12 Wochen.
. Die intravenöse IgG-Infusion ist eine neue und wirksame Methode zur Erhöhung der Thrombozytenzahl während eines akuten Anfalls. ATs blockieren Fc-Rezeptoren von Phagozyten, die eine wichtige Rolle bei blutplättchenhemmenden zytotoxischen Reaktionen spielen; Diese Methode hat als präoperative Vorbereitung für Patienten mit ITP, die einen chirurgischen Eingriff benötigen, an Popularität gewonnen. Eine neue Methode zur Behandlung refraktärer ITP, die zu ermutigenden vorläufigen Ergebnissen geführt hat, ist die Plasmapherese durch eine Säule mit Staphylokokkenproteinen.
Chirurgische Behandlung. Bei der chronischen Form mit starken Blutungen und erfolgloser konservativer Therapie ist eine Splenektomie indiziert. Eine Heilung durch Splenektomie gelingt nicht immer. Verschluss von Milzgefäßen.
Überwachung. Im Stadium der klinischen Remission - Kontrolle der Thrombozytenwerte einmal im Monat. Wenn die klinische und hämatologische Remission länger als 5 Jahre anhält, wird der Patient aus dem Register gestrichen.
Empfehlungen. Beobachtung durch einen Hämatologen am Wohnort. Physiotherapeutische Behandlung und Sonnenbestrahlung sind kontraindiziert. Die Verwendung von Acetylsalicylsäure und Carbenicillin ist kontraindiziert. Vorbeugung von Blutungen - Kräutermedizin (Aufgüsse aus Kamille, Brennnessel, Hagebutte) in Kursen von 15 Tagen alle 3 Monate. Auf Sportunterricht und Sport sollte verzichtet werden. Anmeldung der Behinderung bei anhaltender chronischer Erkrankung.
Komplikationen. Blutungen im Zentralnervensystem. Schwere posthämorrhagische Anämie.
Verlauf und Prognose. Bei den meisten Patienten (80–90 %) tritt die Selbstheilung innerhalb von 1–6 Monaten ein. Wenn der Prozess chronisch ist, ist das Behandlungsschema mit Prednisolon ähnlich. Die Sterblichkeitsrate bei ITP liegt unter 1 %. Todesursachen- Blutungen im Zentralnervensystem, schwere posthämorrhagische Anämie.
Die Ermäßigung. ITP – idiopathische thrombozytopenische Purpura
ICD-10. D69.3 Idiopathische thrombozytopenische Purpura